
Periodontal diseases are amongst the most common inflammatory disease in humans. It has been previously accepted that periodontitis is infectious in origin; newer data suggests that while infection with specific periodontal pathogenic bacteria is necessary for periodontitis to develop, infection alone is insufficient for the development of the disease.
Significantly, the role played by the host, particularly with respect to the mounting of a protective inflammatory response, has not received similar attention, greatly influencing most treatment approaches. As a result, most therapies for periodontitis have focused on reduction and/or elimination of ‘specific’ periodontal pathogens, or on the reduction of the overall oral microbial burden. Surprisingly, even when the microbial burden has been successfully achieved following professional debridement and patient oral hygiene improvement, there are many patients who continue to demonstrate signs and symptoms of periodontitis (reflected by the presence of bleeding on probing and ongoing loss of periodontal attachment).
Furthermore, it is also known that although a patient might present with classic signs of moderate to severe periodontitis (such as pocket depths > 5 mm), the actual disease process may actually be in a state of arrest. This patient, if tested for microbial infection, may continue to harbor classic periodontal pathogenic bacteria. Why then, if periodontal disease is principally infectious, do some patients who are ‘infected’ continue to lose periodontal attachment, while others infected with the very same organisms, are stable or exhibit only gingivitis?
As a corollary, why do some patients with plaque/biofilm-induced gingivitis develop periodontitis, while others never seem to progress beyond gingivitis? Why does this latter population not become infected with high loads of periodontal pathogens that would presumably lead to the development of destructive periodontitis?
Transition from Gingivitis to Periodontitis
It is proposed that there might be a cascade of events (Fig. 1) that lead either to gingivitis or periodontitis depending on the host’s innate immune system (see ‘Immunostat’ below) and their microbial load. During the initial stages of oral plaque formation, it can be considered a relatively ‘non-specific’ biofilm. When initially formed, the biofilm does not contain the classic periodontal pathogenic bacteria. Consequently, a host response is triggered but perhaps only a modest one presenting as gingivitis. If the patient does not smoke, or is not otherwise exposed to periodontal pathogens, and if the host immune response is, as noted above, moderate, it is proposed that gingivitis will not progress to periodontitis.
FIGURE 1. This flowchart demonstrates how inflammation and microbial infection interact with one another. It also explains how periodontitis might develop from gingivitis as a consequence of changes in the local inflammatory milieu and subsequent population with
periodontal pathogenic bacteria. The latter upregulate further, the immune response. In patients with a sensitive immunostat,
which plays the most important role in the box indicating ‘host inflammatory response’.
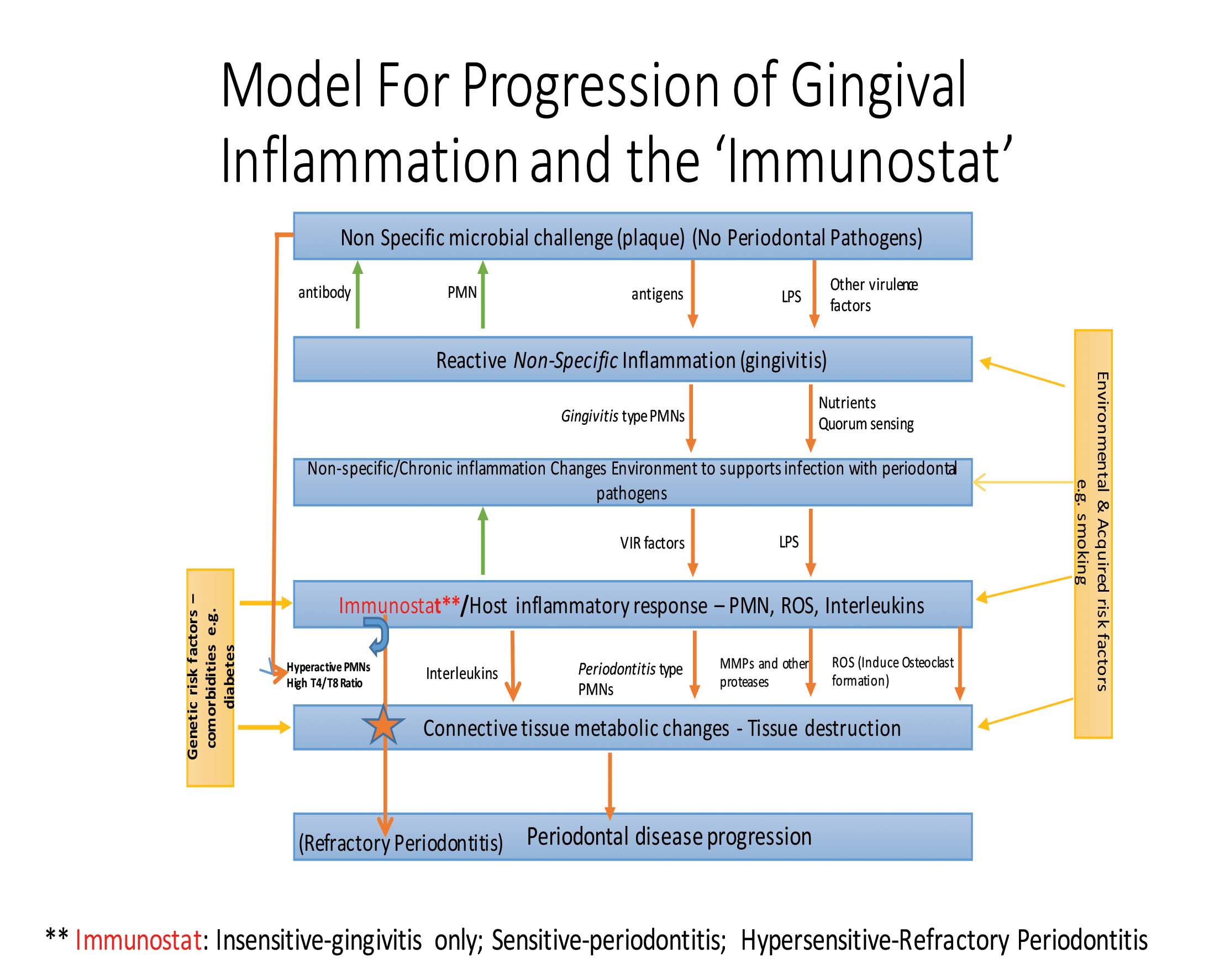
Where the host response is a more vigorous immune reaction, mounted even when the challenge is a relatively non-specific and otherwise pathogen-free biofilm, the biofilm and sulcular environment can be altered (increased levels of GCF, growth factors, chemo-attractants). This new environment may allow periodontal pathogens to thrive in, and/or to be attracted to the biofilm.
As the periodontal pathogens increase, the host immune response may be further stimulated, leading to a dramatic increase in levels of phenotypically distinct or periodontitis-specific polymorphonuclear neutrophils (PMNs). These produce reactive oxygen species (ROS), thereby allowing for the differentiation of osteoclasts, inflammatory modulators, and the production of several proteolytic enzymes (e.g. matrix metalloproteinases or MMPs), which increase inflammation even more. These changes lead to destruction of the surrounding periodontium. This response is magnified in smokers as the aryl hydrocarbons in smoke bind to and activate aryl hydrocarbon receptors, which likely cause differentiation of inflammation-specific PMNs. This would lead to increases in the production and activity of MMPs, as well increased release of ROS and inflammatory cytokines. This cascade of events can be encapsulated by the term ‘oral inflammatory load’ or OIL, and is related directly to loss of clinical attachment.
The increase in OIL, and, as described below, the phenotypic changes in PMNs, are very likely central to the progression of gingivitis to periodontitis, including the progression of the tissue loss observed in patients with periodontitis. Based on the above, the changes in OIL are not related merely to the overall load of oral PMNs, but also to the development of disease specific PMN phenotypes (orally and peripherally). Moreover, the sensitivity of a patient’s innate immune system also determines whether or not periodontitis will be initiated and/or progress; this is the concept of the Immunostat.
Oral Inflammatory Load
The authors have demonstrated that periodontal inflammation can be measured and quantified by PMN levels in oral rinses.1 This oral inflammatory PMN load is a raw measure of inflammation and has also been shown to correlate well with periodontal disease severity.2 A screening test has been developed which enables clinicians and patients to visually ascertain the level of OIL by colorimetric testing. Importantly, this type of monitoring is readily performed in succeeding appointments, thus permitting clinicians and patients to monitor periodontitis activity from the perspective of inflammation levels, measuring, for the first time, reduced inflammation as treatment progresses.
Immunostat
Two distinct para-inflammatory PMN phenotypes are noted in health (Para1 and Para2). Patients with periodontitis exhibit the single hyper-activated pro-inflammatory PMN (Pro).3 The Para1 population is quiescent with few markers of activation and with only a modest ability to produce ROS. The Para2 population is in an intermediary state between Para1 and Pro. Preliminary data suggests that the ratio of the Para1 to Para2 may be indicative of the patient’s immune surveillance state and their likelihood of initiating a frank inflammatory response. The increase in Para2 pushes the immune system closer to an energetic inflammatory response. In the future, this may help to assess a patient’s oral immune status. The sensitivity of an individual’s immune response or their ability to mount an inflammatory reaction can be referred to as the immunostat.
The authors propose that those with a relatively insensitive immunostat are less likely to develop destructive periodontitis. Those with a more sensitive immunostat are predisposed to the development of a strong inflammatory response and subsequent breakdown of periodontal tissue, even with minimal microbiological stimulation. By understanding a priori the characteristics of a patient’s immunostat, it will be possible to develop better and more individualized treatments for patients, accurately predicting treatment outcome or prognosis.
Can immunostat sensitivity be utilized to understand periodontitis in greater detail? Refractory periodontitis presents an excellent model with patients who have hypersensitive immunostats requiring very different treatment (such as host-modulation therapy) as compared to patients with chronic periodontitis.
Refractory Periodontal Disease as a Model for Patients with a Hypersensitive Immunostat
Refractory periodontal disease is the continued loss of periodontal attachment and bone that occurs despite adequate sanative, surgical, and maintenance therapy. Refractory periodontitis was once characterized as a unique disease entity. However, a newer system of disease classification has repositioned refractory periodontitis within a diagnostic category that includes chronic and aggressive periodontitis. In most cases, the condition progresses slowly, often not detected for several years while the patient is undergoing conventional periodontal maintenance therapy. This condition can lead to both functional and aesthetic deficits. For cases where the bone loss has necessitated tooth extraction, implant therapy has been considered. However, it has also been observed that patients who have suffered refractory periodontitis exhibit a higher implant failure rate than patients with severe but chronic periodontitis.4
A number of mechanisms have been proposed to explain the etiology of refractory periodontal disease, including the presence of unique bacteria, or the failure to treat the disease ‘adequately’. However, Bhide et al5 have suggested that the bacterial component may not have as great an influence on the development of disease as was once believed, and that the observation of elevated levels of T-cells (CD4+, CD8+ receptor type) may be characteristic of a “hyper-inflammatory” state in these patients. Additionally, Johnstone et al6 proposed that PMNs from patients with refractory periodontal disease displayed a significantly increased capacity for the production of ROS when compared to PMNs derived from patients with chronic periodontitis. It is probable that both mechanisms may be involved in creating the hyper-inflammatory state that leads to progressive bone and soft tissue destruction.
T-helper cells express the CD4+ surface protein, which plays an active role in cytokine production and the recruitment of other cells. All contribute to the immuno-inflammatory response. T-helper cells produce interleukin -2 (IL-2), interleukin -4 and interleukin -10, along with tumor necrosis factor-B (TNF-B). These factors contribute to the metabolic changes that ultimately lead to connective tissue destruction.
Taken together the above findings suggest that patients with refractory periodontitis may be good exemplars of individuals with a very sensitive immunostat.
Conclusion
Based on the findings discussed above, the authors suggest that periodontitis is not strictly an infectious disease. Periodontitis is an inflammatory disease modulated by microbial, epigenetic, and genetic factors, including the sensitivity of the immunostat. It follows that one can monitor the activity and progression of the disease as well as its prognosis (which is related to progression) by the assessment of OIL as well as identification of phenotypic changes in PMNs (peripheral and oral). Moreover, treatment approaches can and must be developed that target not only the microbial elements of periodontitis but also target the regulation of the host response. This leads to more effective treatment for all forms of periodontitis (e.g. chronic, refractory). Furthermore, by using periodontitis as a model system for inflammatory disease, it is suggested that more effective treatments for other non-oral inflammatory diseases may be developed.
H. C. Tenenbaum, DDS, Dip. Perio., PhD, FRCD(C) is Dentist-in-Chief, Sinai Health System, Professor of Periodontology at the Faculty of Dentistry, and Professor of Laboratory Medicine and Pathobiology, Faculty of Medicine, Mount Sinai Hospital, University of Toronto.
Michael Glogauer, DDS, Dip. Perio., PhD is Professor of Periodontology and a member of the Matrix Dynamics Group at the Faculty of Dentistry.
Michael B. Goldberg, MSc, DDS, Dip. Perio is an Associate Professor in the Discipline of Periodontology at the Faculty of Dentistry, University of Toronto as well as Head of the Division of Periodontology at Mount Sinai Hospital.
Oral Health welcomes this original article.
References:
1. Landzberg M, Doering H, Aboodi GM et al. Quantifying oral inflammatory load: oral neutrophil counts in periodontal health and disease. J Periodontal Res 2015; 50(3):330-6.
2. Bender JS, Thang H, Glogauer M. Novel rinse assay for the quantification of oral neutrophils and the monitoring of chronic periodontal disease. J Periodontal Res 2006; 41:214-20.
3. Fine N, Hassanpour S, Borestein A et al. Distinct Oral Neutrophil Subsets Define Health and Periodontal Disease States. J Dent Res 2016; in-press.
4. Fardal Ø, Linden GJ. Tooth loss and implant outcomes in patients refractory to treatment in a periodontal practice. J Clin Periodontol 2008; 35(8):733-738.
5. Bhide VM, Tenenbaum HC, Goldberg MB. Characterization of Patients presenting for Treatment to a University Refractory Periodontal Disease Unit: Three Case Reports. J Periodontol 2006; 77(2):316-322.
6. Johnstone AM, Koh A, Goldberg MB, Glogauer M. A Hyperactive Neutrophil Phenotype in Patients with Refractory Periodontitis. J Periodontol 2007; 78(9):1788-1794.
Follow the Oral Health Group on Facebook, Instagram, Twitter and LinkedIn for the latest updates on news, clinical articles, practice management and more!











